mesoの実験ノート
GGRNA ver.2リリース
- 2013-08-23 (金)
- DBCLS
7月8日に GGRNA ver.2 をリリースしました。GGRNAを公開してから初のメジャーアップデートになります。少し時間が経ってしまいましたが、この記事では、アップデートによる主な変更点をまとめました。
全生物種に拡張
これまでのGGRNAは、RefSeqに収録された転写産物のうち、13種類のモデル生物に限ってサービスを提供してきました。今回のアップデートでは、これを全生物種に拡張し、しかも同時に検索できる Zoo というモードを用意しました(検索例:CAAGAAGAGATTG )。

ちなみに RefSeq は、ゲノム、mRNA、ncRNA、タンパク質のエントリから構成されており、GGRNAではこのうちmRNA(IDがNM_, XM_で始まる)および ncRNA(NR_, XR_)を検索できます。
GGRNAの元データ(NCBI RefSeq)
ftp://ftp.ncbi.nlm.nih.gov/refseq/release/complete/complete.*.rna.gbff.gz
技術面では、データベースの増大に対応するために、内部の検索エンジンを最新版の Sedue に置き換えました。これまでのGGRNAでは、圧縮接尾辞配列のインデックスをメモリに載せて検索していたため、メモリ容量の制約で13種類のモデル生物しか入りませんでした。また、ヒット件数が極端に多い場合に、圧縮接尾辞配列の解凍処理がネックになりすごく時間がかかるという弱点がありました。最新版のSedueでは(非圧縮の)接尾辞配列のインデックスをSSDに載せることによって、これらの弱点を克服しています。
最近SSDはずいぶん安価になってきて、2013/8/20の時点で960GBのモデルが5.9万円で販売されています(価格.com)。約3TBのスペースを確保すれば、GenBank/EMBL/DDBJ国際塩基配列データベースに登録されたゲノム以外の全てのエントリを、配列を含めて全文検索できるようになります。実はもう手元のテスト環境では動いており、データ更新などいくつかの問題を解決できたら公開する予定です。
REST APIを完備
クエリとURIとを1対1対応させました。検索結果に直接リンクを張ったり、機械的にアクセスして検索結果をテキスト形式やJSONで取得することが、より簡単にできるようになりました。
URI
http://GGRNA.dbcls.jp/species/query+string[.format][.download]species → 生物種。hs (ヒト), mm (マウス), dm (ハエ), …。省略時はヒト。
query+string → 検索ワード。特殊文字はURIエンコードすること。
format → html, txt, json。省略時はhtml。
download → ファイルとしてダウンロード。詳細は GGRNA.v2 REST API仕様 を参照。
検索例:
- http://GGRNA.dbcls.jp/NM_001518
→「NM_001518」を検索。.format の部分を省略した場合は html になる。 - http://GGRNA.dbcls.jp/hs/homeobox.txt
→ ヒトで「homeobox」を検索してタブ区切りテキストで取得。 - http://GGRNA.dbcls.jp/dm/%22RNA+interference%22.json
→ ショウジョウバエで「”RNA interference”」をフレーズ検索してJSONで取得。
URIでは ” は %22、スペースは + に置換する(URIエンコード)。 - http://GGRNA.dbcls.jp/ce/caagaagagattg.txt.download
→ 線虫で「caagaagagattg」を検索してタブ区切りテキストをダウンロード。
なお、旧バージョンで提供してきたREST APIもそのまま残しており、以前と同じURIで情報を取得できますが、
http://GGRNA.dbcls.jp/api/* → http://GGRNA.dbcls.jp/v1/api/*
にリダイレクトされる点はご注意ください。
マイクロアレイのプローブ情報を網羅的に収録
GGRNAでマイクロアレイのプローブIDを検索すると(例:1552311_a_at )、自動的にプローブの配列に変換したうえでその結合部位を検索する機能があります。今回のアップデートでGGRNAはあらゆる生物種に対応したので、それに合わせてマイクロアレイのプローブ情報も追加しました。詳細は別の記事に書きますが、AffymetrixとAgilentのほぼ全てのマイクロアレイ製品のプローブIDと配列を網羅的に収録しています。
検索結果のランキングを改良
遺伝子名のつもりで Dicerを検索 しても、アミノ酸配列のなかの “DICER” ばかり上位に表示されて残念だったので、キーワードがヒットした場所に応じて重みを付けることにしました。具体的には、遺伝子名やタイトルにマッチした場合は相対的に高いスコアを、アミノ酸配列や塩基配列にマッチした場合は相対的に低いスコアを与えています。
ランキングは今後さらに改良していく必要があり、たとえばメジャーな生物種をより上位に表示したり、Accession番号が XM_/XR_ で始まる “PREDICTED” な配列のスコアを相対的に下げたりすると使いやすくなりそうです。ただし、なるべく多くのユーザの意図に沿うバランスの良いスコアリングを慎重に検討する必要があります。また、GGRNAのランキングがユーザの意図に沿っていなかった場合でも(たとえば本当に “DICER” というアミノ酸配列を探したかった場合でも)、ファセットナビゲーションのような仕組みで即座に目的のものを絞り込めるようにしたいです。
塩基配列のミスマッチ検索を廃止 → GGGenomeに移行
GGRNAの内部エンジンを変更したことにより、ミスマッチを含む塩基配列の検索ができなくなりました。配列検索にこだわってきたGGRNAとしては、このダウングレードは痛手なのですが、データベースの拡張性を優先してミスマッチ検索の廃止を決めました。そのかわりに、塩基配列の検索に特化した GGGenome《ゲゲゲノム》というサイトを新たに立ち上げ、ミスマッチやギャップを含む塩基配列の検索はそちらに引き継ぐことにしました。
高速塩基配列検索 GGGenome《ゲゲゲノム》
http://GGGenome.dbcls.jp/
GGGenomeでミスマッチやギャップを含む塩基配列を検索した例:
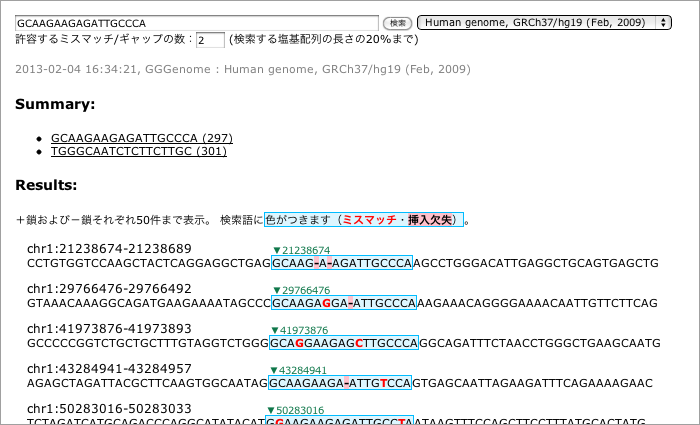
Gene OntologyおよびEC番号検索の停止
旧バージョンでは、Gene OntologyのIDやterm、酵素EC番号の情報をRefSeqの転写産物に紐付けることによって、GGRNAで検索できるようにしていました。しかし、この機能も内部エンジンの更新に伴いしばらく提供できなくなります。こちらは再開に向けて開発を進めます。
ソースコード公開
今回のアップデートを機にソースコードを公開しました。
ソースコード(GitHub)
https://github.com/meso-cacase/GGRNA
内部エンジンに使用している Sedue は商用ソフトウェアでソースは公開されていませんが、プリファードインフラストラクチャー(PFI)から入手が可能です。Sedueでは、接尾辞配列のインデックスをSSDに保持することによって、塩基配列やアミノ酸配列を含む検索ワードを、見落としなく、きわめて高速に検索しています。
旧バージョン
今回のアップデートでは大きく変更した部分や削除した機能もあるので、旧バージョンもしばらく提供します。ただし、RefSeqのリリースに合わせた2ヶ月に一度のデータベース更新はせず、2013年7月のRefSeq release 60で更新を終了します。
GGRNA ver.1
http://GGRNA.dbcls.jp/v1/
- Comments (Close): 0
- Trackbacks: 0
BioHackathon 2013でやりたいこと
- 2013-06-25 (火)
- DBCLS
6/24(月)〜28(金)はDBCLSにて国際開発者会議 BioHackathon 2013 が行われています。この期間にmesoがやりたいことをまとめました。
【GGRNA】GGRNA 2.0 リリース&ソース公開
現在の GGRNA は、圧縮接尾辞配列のインデックスをメモリに載せて検索しているため、メモリ容量の制約から13種類のモデル生物に限ってサービスを提供してきました。また、ヒット件数が極端に多い場合は圧縮接尾辞配列の解凍にすごく時間がかかるという弱点がありました。そこで、内部の検索エンジンを最新版のSedueに置き換えて、(非圧縮の)接尾辞配列のインデックスをSSDに載せる方針(以下、SSD方式)に変更することを計画しています。
SSD方式のメリットは、データベースの容量の制約がなくなることです。たとえば約3TBのSSD領域を用意すれば、GenBank/EMBL/DDBJ国際塩基配列データベースに登録されたゲノム以外の全ての配列を全文検索できるようになります(実はもうやっていてテスト段階)。また、ヒット件数が極端に多い場合でも素早く結果を返せるようになります。
逆にデメリットは、現在のGGRNAで提供している機能のうち、ミスマッチを含む塩基配列の検索ができなくなることと、塩基配列やアミノ酸配列の hit position が取得できなくなること。配列検索にこだわっているGGRNAとしては、このダウングレードは痛手なので、なかなかSSD方式への移行を決断できませんでした。
いろいろと悩んだ結果、ミスマッチを含む塩基配列の検索は GGGenome《ゲゲゲノム》という配列検索に特化したサービスに移してしまうことにしました。また、PFI社の大野さんをはじめとするメンバーがSedueのhit position取得プラグインを作成中で、これが完成すればSSD方式に移行できそうです。
7月には統合データベースのユーザ評価が比較的大規模に行われる予定で、その対象サービスのひとつにGGRNAを入れて頂いたので、多くの方のフィードバックをいただけるチャンスです。それまでにSSD方式への移行を済ませて GGRNA 2.0 をリリースしたいと考えています。
さらに、GGRNA 2.0 からはソースを公開する予定です。実はGGRNAの論文を査読してくださったreviewerのひとりが、オープンソースでやったらどうかとコメントしてくれたのですが、ぜひそうしたいと返答してからかなり時間が経ってしまいました(やるやるサギ)。このreviewerはたぶん国内の知っている方で、とても丁寧に査読をしてくれて多くのconstructiveなコメントを頂き感動したのですが、そのときの約束を守れていないので顔向けできない状況なのです(笑
(8/26更新)
7/8に GGRNA ver.2をリリース しました。
【GGRNA/GGGenome】Galaxyと連携
GGRNA / GGGenome の基本的な検索機能はほぼ完成しているので、他のツールとの連携を強化したいと思っていたところ、@32nm さんからこんな連絡をいただきました。
@meso_cacase GGRNA にはGalaxy のGet Data/UCSC Main table browserのように検索結果をSend query to Galaxyする機能を実装する予定はありすか?特に配列検索でData Exportのタブ切り相当で返るように (Twitter: 2013/5/29 – 0:33)
それは良さそうですね、と返信すると参考文献を教えてくださいました。
@meso_cacase どもども。http://database.oxfordjournals.org/content/2011/bar011.full が参考になるはずです〜 (Twitter: 2013/5/29 – 22:42)
【GGGenome】検索結果のうち特定のフィールドだけを返すREST API
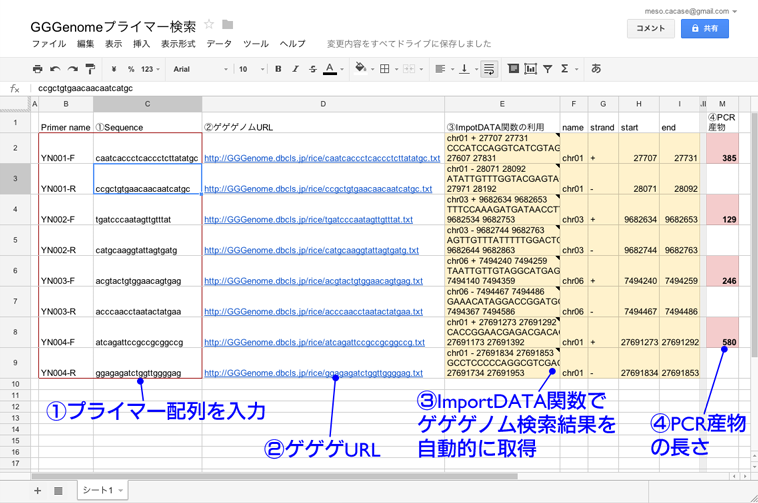
GGGenome は検索クエリがURLと1対1対応しており、しかもURLの末尾に .txt をつけると検索結果をタブ区切りテキストで取得できます(例: http://GGGenome.dbcls.jp/TTCATTGACAACATT.txt)。この機能を利用すると、スプレッドシートにプライマー配列をコピペするだけで、自動的にGGGenome検索をしてPCR産物の長さや増幅する場所を求めることができます(図)。このとき、GGGenomeの検索結果のうち特定のフィールド(例えば start position)だけを取得できる手段があるとさらに便利なので、TogoWS のURL規則を参考に実装したいです。
下の図では、Google Driveの関数 ImportDATA( URL ) を利用してGGGenomeの結果を直接セルにインポートしてみました。「①プライマー配列を入力」のところだけ入力すると、残りの部分は自動的に埋まります。2つのプライマー間で染色体上の座標を引き算すれば、PCR産物の長さも計算できます。
(6/27更新)
上記の Googleスプレッドシート を読み出し専用で公開しました。ImportDATA 関数によるGoogleスプレッドシートとGGGenomeの連携法は、@hkanekane さんから教えていただきました。
【GGGenome】TogoGenomeと連携して遺伝子名表示
GGGenome で塩基配列を検索すると染色体上の座標が返ってきますが、TogoGenome と連携してその位置にある遺伝子名なども表示できれば便利になるはず。
【BioRuby】siRNA設計ライブラリのアップデート
BioRubyには二階堂さん作成のsiRNA設計ライブラリ(sirna.rb)があり、そのなかで Ui-Teiらの設計法とReynoldsらの設計法(リンク先のFig. 1-1を参照)が実装されています。mesoは前者を発表したチームの一人で(Ui-Tei et al., Nucleic Acids Res. 2004)、sirna.rb のコードを読んでみると各ruleの解釈が原著論文の意図と少し違う部分があることに気づいたので、下記のようにアップデートのリクエストをしようと思っています:
ハッカソンではBioRubyのメジャーアップデートが予定されているそうなので、それにあわせて採用してもらえるとありがたいです。
(6/27更新)
ユニットテスト(test_sirna.rb)も修正し、コミットメッセージを詳しく書いてpull requestすればよいとのこと。
(8/26更新)
下記のように、pull requestを取り込んでいただきました。
- Comments (Close): 0
- Trackbacks: 0
difff《デュフフ》リニューアル
- 2013-03-13 (水)
- DBCLS
テキスト比較ツール difff《デュフフ》をリニューアルしました。
- difff《デュフフ》ver.6 : http://altair.dbcls.jp/difff/
difff《デュフフ》はwebベースのテキスト比較ツールとしてmesoが9年前にこっそり作ったプログラムで、下の画像のように2つのテキストの差分(diff)を示してくれます。もともとはdifffと書いてディフ・エフと読んでいましたが、あるツイートをきっかけに《デュフフ》と呼ばれるようになりました。

なお、旧バージョン (ver.5) もしばらく残しておきます。
今回の変更点
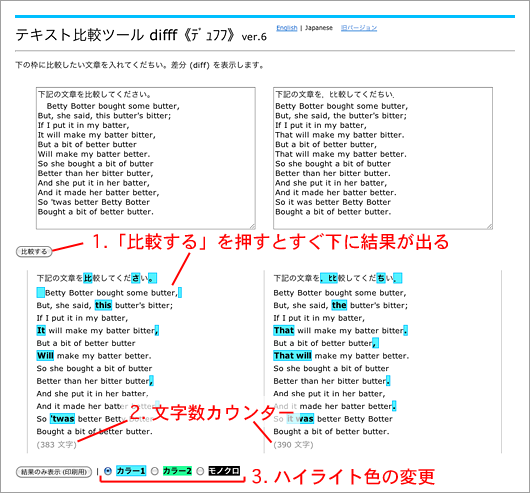
1. 入力フォームのすぐ下に結果を表示
今回のバージョンから、入力フォームのすぐ下に結果が表示されます。結果を見ながらフォームのテキストを修正し、すぐにまた デュフフ できるようになりました。
この変更により、もとのテキストと比較結果をまとめて1つのHTMLファイルに保存できるようになりました。しかも、保存したHTMLファイルを開けば デュフフ の作業を再開できます。(ただしGoogle Chromeでは保存時に「ウェブページ、完全」を選ばないとうまく保存されず、またこの方法で保存したファイルから デュフフ を再開できないようです。)
印刷するときなど入力フォームが邪魔なときは「結果のみ表示 (印刷用)」というボタンを押せばフォームを隠すことができます。
2. 文字数カウンター
入力されたテキストの文字数をカウントする機能をつけました。現状では length() を取っているだけなので改行・スペースも1文字としてカウントされます。しかし一般的には改行・スペースを数えないか、スペースだけ数えることが多いようなので、少し調べてアップデートする予定です(ついでに英文の単語数も表示すると便利かも)。
(3/22更新)いろいろな要望があるようなので下記のように全部表示することにしました。ちなみにハイライトは3/21から季節限定で桜色になっています。ここでいう空白とは、半角スペース、全角スペース、タブです。単語数は、空白/改行によって隔てられた、空白/改行以外のまとまりの個数です(テキストに対して s/\s*\S+//g が成功した回数です)。

3. ハイライト色の変更
今までの蛍光っぽいグリーンは長文だと目がチラチラと疲れるので、ソフトな水色系に変更しました。まわりを少し濃い色で囲って目立つようにしています。今までの蛍光グリーンに慣れている人は、蛍光グリーンにも切り替えられます。また、印刷に適した白黒表示に切り替えることもできます。
ちなみに一般公開していなかった頃のdifffは赤文字+蛍光イエローで、書体も明朝/ローマンでした。

4. プログラム内部の日本語処理をUTF-8に変更
初代difffを書いた当時、Perlで日本語を扱うときはEUC-JPでコーディングするのが一般的でした。difffでも、コードと入出力をEUC-JPに統一したうえで、EUC-JPの1文字にマッチする正規表現を使い、文字ごとの差分を検出していました。
# EUC-JPの1文字にマッチする正規表現(今となっては懐かしのコード)
my $ascii = ‘[\x00-\x7F]’ ; # ASCII文字の集合
my $twobyte = ‘(?:[\x8E\xA1-\xFE][\xA1-\xFE])’ ; # 2バイトEUC-JP文字の集合
my $threebyte = ‘(?:\x8F[\xA1-\xFE][\xA1-\xFE])’ ; # 3バイトEUC-JP文字の集合
my $eucjp = “(?:$ascii|$twobyte|$threebyte)” ; # EUC-JP文字全体の集合
現在のPerlでは、UTF-8でコーディングすれば簡単に日本語を扱うことができます(参考 : Perl5.8でUTF-8のメモ / 楽)。たとえば、正規表現の /./ が日本語の1文字にマッチし、length(‘ほげほげ’) で4が返ってきます。difffも今回のリニューアルを期に(やっと)UTF-8に移行しました。
ソースコード
mesoが管理している difff《デュフフ》のウェブサイトは誰でも無償で利用でき、入力テキストも一切サーバに残らないようになっています。しかし、機密文書をどうしても社内のサーバで デュフフ したいという声もあったため、GitHubでソースを公開しています。どうぞご利用ください。
- GitHub : meso-cacase / difff
- Comments (Close): 0
- Trackbacks: 0
DDBJ 89.0 (Jun, 2012)
- 2012-08-10 (金)
- DBCLS
DDBJのデータベースについて記事を書いた直後に89.0がリリースされたのでメモ。無脊椎 (INV) はエントリ数、塩基数とも減っていた。理由を調べてみると、生物種がはっきり書かれていないエントリなどが大量にremoveされていた(例:GU672000)。
一通りダウンロードして、現在は塩基配列部分をインデクシング中。
| Division | 内容 | エントリ | 塩基 | ファイルサイズ | (GB) |
|---|---|---|---|---|---|
| 合計 | 160,254,629 | 141,016,380,296 | 561,920,288,622 | 523.3 | |
| HUM | ヒト | 563,386 | 4,996,058,095 | 8,374,749,482 | 7.8 |
| PRI | 霊長類 (ヒトを除く) | 102,989 | 1,292,811,012 | 1,891,443,366 | 1.8 |
| ROD | 齧歯類 | 440,159 | 4,430,778,988 | 7,005,892,095 | 6.5 |
| MAM | 哺乳類 (ヒト,霊長類,齧歯類を除く) | 312,768 | 852,651,001 | 1,781,940,910 | 1.7 |
| VRT | 脊椎動物 (ヒト,霊長類,齧歯類,哺乳類を除く) | 1,057,240 | 2,845,002,278 | 6,069,241,055 | 5.7 |
| INV | 無脊椎動物 | 1,577,829 | 2,443,533,451 | 6,843,835,702 | 6.4 |
| PLN | 植物・真菌類 など | 2,471,569 | 5,840,633,857 | 13,652,436,941 | 12.7 |
| BCT | バクテリア | 841,541 | 8,158,523,792 | 19,074,978,516 | 17.8 |
| VRL | ウイルス | 1,185,860 | 1,371,199,251 | 4,914,556,954 | 4.6 |
| PHG | バクテリオファージ | 6,512 | 77,382,875 | 187,608,552 | 0.2 |
| PAT | 特許出願に含まれる塩基配列データ | 23,998,516 | 11,945,459,364 | 42,322,850,200 | 39.4 |
| ENV | PCR,DGGE,あるいは,その他の方法で直接, 分子を単離した環境上のサンプルに由来した配列 | 4,715,129 | 3,237,988,584 | 12,653,785,921 | 11.8 |
| SYN | synthetic constructs 人為的に構成された配列 | 123,112 | 926,662,970 | 1,643,396,741 | 1.5 |
| EST | expressed sequence tags short single pass の cDNA 配列 | 73,142,305 | 40,637,013,620 | 239,749,294,113 | 223.3 |
| TSA | transcriptome shotgun assemblies 再構成された (assembled) mRNA 配列 | 6,714,908 | 4,679,084,409 | 18,903,720,242 | 17.6 |
| GSS | genome survey sequences short single pass のゲノム配列 | 33,998,784 | 21,641,585,155 | 99,772,519,365 | 92.9 |
| HTC | high throughput cDNA sequences EST 以外の大規模 cDNA 配列プロジェクトに由来。 最終的に,生物種による division に移される場合がある。 | 551,351 | 634,629,961 | 2,512,724,377 | 2.3 |
| HTG | high throughput genomic sequences ゲノムプロジェクトに由来。 最終的に,生物種による division に移される。 | 146,427 | 24,368,642,654 | 32,103,302,381 | 29.9 |
| STS | sequence tagged sites Genome sequencing の tag となる配列。 chromosome, map, PCR_condition 等の情報が必要。 | 1,322,639 | 636,259,470 | 4,497,106,490 | 4.2 |
| UNA | 未注釈データ 最近は UNA division は使用していない。 | 290 | 479,509 | 1,379,737 | 0.001 |
| CON | Contig / Constructed ゲノムプロジェクトのように個々に登録された一連の 配列データを結合し,1つのアクセッション番号を付与 した長大なデータ。塩基配列は記載されない。 | 6,981,315 | 0 | 37,963,525,482 | 35.4 |
※ 表はDDBJ 89.0の リリースノート を集計。Divisionの説明は「DDBJ のデータ公開形式 (flat file) の説明」を抜粋。
- Comments (Close): 0
- Trackbacks: 0
マイクロアレイ学生実習
- 2012-06-28 (木)
- DBCLS
6/12-20の期間、東京大学理学部生物化学科・生物情報科学科3年生の学生実習を担当しました。テーマは「マイクロアレイを用いた網羅的遺伝子発現解析」。生物情報では、2009年にAgilent社のマイクロアレイ設備一式を学生実習のために導入しており、学科の共通機器として毎年の学生実習に使用しているほか、各ラボの研究にも活用されています。調達時にAffymetrixとAgilentの方に伺ったところ、学部の学生実習でマイクロアレイをやるところは聞いたことがないとのこと。実習のプロトコルも試行錯誤を重ね、今年で4年目になります。
miRNAによる標的遺伝子の抑制
実習では、ヒトのHeLa細胞にマイクロRNA (miRNA) を導入して24時間後に回収したものを各班に配布。miRNA導入による遺伝子発現の変動を、マイクロアレイで調べるのがねらいです。
miRNAは、5′末端から2-8の位置の ‘seed’ と呼ばれる領域をたよりにmRNAの3′ UTRと結合し、翻訳抑制やmRNAの分解を誘導することが知られています。
ヒトのlet-7b:5′-UGAGGUAGUAGGUUGUGUGGUU-3′ (miRBaseより。赤字部分がseed)
実習で使用したmiRNAとは異なりますが、たとえばヒトのmiRNAのひとつであるlet-7bの場合は、赤字で示した GAGGUAG がseedと呼ばれる領域です。これと相補的な CUACCUC という配列を3′ UTRにもつ (以下、seedマッチする と表現) mRNAは、let-7bの標的となりその発現が抑制される可能性があります。ただし、seedマッチするmRNAのすべてが標的となるわけではなく、またseedマッチ以外にもさまざまなfactorが標的の認識および抑制に寄与することがわかっています。miRNAの標的予測については、TargetScan、PicTar、miRandaなどさまざまなプログラムが開発されていますが、いずれもfalse positiveが多く、正確な予測は難しいのが現状です。
seedマッチする遺伝子の変動
実習では1日目に細胞からtotal RNAを抽出、2日目に逆転写とcRNA合成、3日目にアレイへのハイブリダイゼーション、4日目にアレイのスキャンをおこない、4〜6日目の3日間でデータの解析をしました。この3日間は情報基盤センターの演習室をお借りしました。
データの解析には、以下のソフトを利用しました。
- Excel – マイクロアレイのデータ解析をブラックボックスにしないために、まずは生の数値にきちんと触れてもらいました。アレイ間の正規化をおこない各種プロットを作成。
- GeneSpring GX – エクセルで全部やるのは苦行なので、一連のデータ解析は基本的にGeneSpring GXを使用。Agilent社より学生実習のための期間限定ライセンスを供与いただきました。
- R – 今年はスムーズに進んだのでRによる解析も紹介。マイクロアレイデータ解析用のパッケージlimmaを利用し、コードの基本部分は講師が提供。
細胞に導入したmiRNAとseedマッチする遺伝子をリストアップして、それらの変動のようすをMAプロット上に可視化してみます。すると、seedマッチする遺伝子群 (青色) は、その他の遺伝子群 (水色) と比較して下側に分布しており、発現量が減少している傾向が確認できました。ただし減少というのはあくまで集団としての傾向で、個々の遺伝子に注目すると、seedマッチしていても変化がないか、増加しているような遺伝子もたくさんあります。

発現量が減少している傾向を、より定量的に評価してみます。seedマッチする遺伝子群 (上のMAプロットで青色)、その他の遺伝子群 (水色) のそれぞれについて、MAプロットの縦軸の値「発現量の変動」でソートしたときの累積度数を求め、プロットします。

その他の遺伝子群 (黒色の線) は、(0, 0.5) を通る対称的な曲線になりました。一方、seedマッチする遺伝子群 (青色の線) は、それよりも左方向にシフトしており、発現量が減少している傾向がはっきりわかります。もしまったく減少していなければ、黒い曲線と重なるはずです。このように、MAプロットでは減少がわかりにくい場合でも、累積度数曲線を作成すると評価しやすくなります。
miRNAのseedが修飾されると標的は変わるか?
今回の実習に使用したmiRNAは、seed領域のAがI (イノシン) に修飾されることが知られています。このときmiRNAの標的となる遺伝子群はどのように変化するのでしょうか。実習では、AをGに置換したmiRNA (G-type)、および、AをIに置換したmiRNA (I-type) についてもマイクロアレイをおこないました。
G-type を導入した細胞では、もともとのseedがマッチしていた遺伝子群は抑制されず、A → G に置換したseedがマッチする遺伝子群が抑制されるようになりました。seedが1塩基かわるだけで、標的となる遺伝子群が大きく変わってしまうことを実感できたのではないかと思います。
一方、I-type を導入した細胞では、やはりもともとのseedがマッチしていた遺伝子群は抑制されず、A → G 置換したseedがマッチする遺伝子群が少し抑制されていました。また、A → U 置換したseedがマッチする遺伝子群、A → C 置換したseedがマッチする遺伝子群は、どちらも抑制されていませんでした。
イノシンは複数種類の塩基 (C, A, U) と塩基対を形成できますが、実習の結果を考慮すると、seed領域の A → I 修飾は、A → G 置換に近い効果がありそうです。もちろんこれを一般的に示すには、今回の実習で使用した特定のmiRNAのアレイデータだけでは不十分ですし、レポーター実験などによる詳細な検討も必要でしょう。
もっとも、6日間の学生実習でここまで解析ができて発展性もありそうな結果が出たのはよかったのではないかと思います。
- Comments (Close): 0
- Trackbacks: 0
DDBJの塩基配列データベース
- 2012-06-25 (月)
- DBCLS
GGRNAにGenBank/EMBL/DDBJの塩基配列を全部入れようとしています。塩基配列は3極で毎日交換されており、各FTPサイトから同様のデータセットをダウンロードできるのですが、GenBankよりDDBJのサイトのほうがきっちり整理されていて、説明も丁寧なように見えます(例:DDBJ のデータ公開形式 (flat file) の説明 など日本語の解説も充実)。しかもファイルの転送が高速で、GenBankからダウンロードすると一晩かかるところが、DDBJからだと20分程度で約70GBの圧縮ファイルをダウンロードできます。
データベースは21のdivisionに別れています。簡単なスクリプトを書いて、DDBJ 88.0 (2011年12月) の リリースノート を下記のように集計してみました。展開後のファイルサイズはDDBJ 88.0全体で約500GBありますが、ほとんどのユーザにとっては、上から13個のHUM〜SYN (111GB) あるいはそれにESTを加えたもの (330GB) が検索できれば十分のような気がします。全部をGGRNAに入れると余計なヒットが増える上にスピードも遅くなり、結果として使いにくくなるかも。。。
しかし、まずは全部を入れてみて、使い勝手を評価しつつ良い方法を考えていこうと思います。
| Division | 内容 | エントリ | 塩基 | ファイルサイズ | (GB) |
|---|---|---|---|---|---|
| 合計 | 152,763,469 | 134,956,109,049 | 536,061,081,910 | 499.2 | |
| HUM | ヒト | 549,320 | 4,871,171,790 | 8,108,485,740 | 7.6 |
| PRI | 霊長類 (ヒトを除く) | 100,839 | 1,290,713,207 | 1,884,393,225 | 1.8 |
| ROD | 齧歯類 | 428,928 | 4,415,260,756 | 6,956,941,030 | 6.5 |
| MAM | 哺乳類 (ヒト,霊長類,齧歯類を除く) | 296,080 | 827,310,516 | 1,710,667,122 | 1.6 |
| VRT | 脊椎動物 (ヒト,霊長類,齧歯類,哺乳類を除く) | 901,031 | 2,736,438,170 | 5,651,416,041 | 5.3 |
| INV | 無脊椎動物 | 1,705,900 | 2,490,017,114 | 7,325,302,790 | 6.8 |
| PLN | 植物・真菌類 など | 2,267,506 | 5,552,139,564 | 12,782,926,982 | 11.9 |
| BCT | バクテリア | 766,137 | 7,342,956,895 | 17,261,515,942 | 16.1 |
| VRL | ウイルス | 1,097,112 | 1,252,521,302 | 4,502,279,650 | 4.2 |
| PHG | バクテリオファージ | 6,365 | 69,569,157 | 169,429,217 | 0.2 |
| PAT | 特許出願に含まれる塩基配列データ | 23,134,648 | 11,447,354,630 | 41,298,319,167 | 38.5 |
| ENV | PCR,DGGE,あるいは,その他の方法で直接, 分子を単離した環境上のサンプルに由来した配列 | 3,973,175 | 2,662,200,445 | 10,296,471,578 | 9.6 |
| SYN | synthetic constructs 人為的に構成された配列 | 121,592 | 922,229,249 | 1,633,026,042 | 1.5 |
| EST | expressed sequence tags short single pass の cDNA 配列 | 71,312,541 | 39,638,590,086 | 234,127,930,692 | 218.0 |
| TSA | transcriptome shotgun assemblies 再構成された (assembled) mRNA 配列 | 4,322,705 | 2,821,816,096 | 11,585,871,018 | 10.8 |
| GSS | genome survey sequences short single pass のゲノム配列 | 32,874,011 | 21,009,093,483 | 96,812,686,537 | 90.2 |
| HTC | high throughput cDNA sequences EST 以外の大規模 cDNA 配列プロジェクトに由来。 最終的に,生物種による division に移される場合がある。 | 535,729 | 611,638,933 | 2,441,930,865 | 2.3 |
| HTG | high throughput genomic sequences ゲノムプロジェクトに由来。 最終的に,生物種による division に移される。 | 145,891 | 24,358,635,476 | 32,097,295,008 | 29.9 |
| STS | sequence tagged sites Genome sequencing の tag となる配列。 chromosome, map, PCR_condition 等の情報が必要。 | 1,322,165 | 635,972,107 | 4,496,495,798 | 4.2 |
| UNA | 未注釈データ 最近は UNA division は使用していない。 | 290 | 480,073 | 1,381,592 | 0.001 |
| CON | Contig / Constructed ゲノムプロジェクトのように個々に登録された一連の 配列データを結合し,1つのアクセッション番号を付与 した長大なデータ。塩基配列は記載されない。 | 6,901,504 | 0 | 34,916,315,874 | 32.5 |
※ 表はDDBJ 88.0の リリースノート を集計。Divisionの説明は「DDBJ のデータ公開形式 (flat file) の説明」を抜粋。
- Comments (Close): 0
- Trackbacks: 0
GGRNAの論文掲載+今後の計画
- 2012-06-05 (火)
- DBCLS
GGRNA論文掲載
GGRNAの論文が5/28付でNucleic Acids Research誌のオンライン版に掲載されました。毎年7/1に発行されるWeb Server Issueに収録されます。
Yuki Naito & Hidemasa Bono (2012)
GGRNA: an ultrafast, transcript-oriented search engine for genes and transcripts.
Nucleic Acids Res. (Web Server Issue) DOI:10.1093/nar/gks448
論文公表にあわせて、日本語の解説を「ライフサイエンス 新着論文レビュー」の番外編として公開しました。
統合遺伝子検索GGRNA:遺伝子をGoogleのように検索できるウェブサーバ.
内藤雄樹・坊農秀雅 (2012) ライフサイエンス 新着論文レビュー・DBCLSからの成果発信
[印刷用PDF]
「ライフサイエンス 新着論文レビュー」はDBCLSが提供している日本語コンテンツのひとつで、最近公開された日本人を著者とする生命科学分野の論文を、著者自身がわかりやすく紹介・解説したものです。Nucleic Acids Researchは残念ながら新着論文レビューの掲載対象誌ではありませんが、DBCLSからの成果を広く発信するために、番外編として記事を執筆させてもらえることになりました。
まずは編集長の飯田さんに草稿を見ていただきました。するとその日のうちに校正が。これはすごい。とても読みやすくなりました。平仮名にする単語、句読点のつかい方、単語の順序などに気をつけるだけでも、かなり読みやすくなるようです。もちろんそれだけでなく、大幅に手を加えていただいた箇所もあります。
ビフォーとアフターを、difff《デュフフ》を使って表示してみると、文章が美しくなっている様子がわかります。

difff《デュフフ》は文書作成支援ツールとしてmesoが8年前にこっそり作ったプログラムです。誰でも使えるように4月に公開したところ、GIGAZINEで紹介されて話題に。たぶん @h_ono さんがつけてくれた名前が素敵だったからでしょう。デュフフ
さて、GGRNAの論文化にあたり、現状の問題点や今後やりたいことが見えてきたので、まとめてみたいと思います。
今後の課題と開発計画
ヒット数が多い場合の高速化
現在のGGRNAにはひとつ問題が残っています。ヒット件数が極端に多いと、圧縮suffix arrayの解凍がネックになりひじょうに時間がかかるのです。たとえば「cancer」「aaaa」を検索するとしばらく結果が返ってきません(しかもヒットが多すぎるため途中で検索が打ち切られます)。また「”translation initiation factor”」というフレーズをダブルクオートをつけずに検索すると、「translation」「initiation」「factor」を別々に検索して結果の共通部分をとるので、ひじょうに時間がかかるうえに意図した結果が返ってきません。これらの問題は、実際にGGRNAを使ってみると頻繁に起こるため、早急に何とかしたいところです。
GGRNAでは内部のエンジンとしてPFIのSedue FlexとMySQLを組み合わせて検索を実行していますが、これらをSSDベースの新しいSedueに置き換えることを計画しています。新しいSedueはsuffix arrayのインデックスを圧縮せずSSDに載せる方式で、大量にヒットした場合でもインデックスの解凍が不要なためかなりの高速化が期待できます。予備的な検討では、前述のようなクエリを含めほとんどが2, 3秒以内に検索できています。ただしエンジンを載せ替えるためにはGGRNAのコードのかなりの部分を書き換えなければなりません。ちょっと時間がかかりそうです。
RefSeq全種+国際塩基配列DB対応
内部のエンジンをSSDベースの新しいSedueに置き換えることによって、データベースの大きさの制約が大幅に緩和されます。現在512GBのSSDは4万円程度まで価格が下がっており (→ 価格.com)、これに50〜100GBのデータベースが載ります。サーバ用のSSDが割高であることを考慮しても、数百GB程度のデータベースであればGGRNAに載せることが十分に可能です。
現在のGGRNAは、RefSeqのうち13種のモデル生物に対応していますが、今年度中にRefSeqの全生物種+GenBank/EMBL/DDBJ国際塩基配列データベース(INSDC)に登録されたゲノム以外の配列全体を取り込んでしまおうと計画しています。データベースの大きさは、現在のGGRNAの約100倍になる見込みです。
参考:
- RefSeq mRNA 13生物種.gz:0.7GB(解凍後約4GB)
- RefSeq mRNA全体.gz:4.0GB(解凍後約20GB)
- INSDCゲノム以外.gz:64.4GB(解凍後約350GB)
また、INSDCを構成する機関のひとつであるDDBJは、DBCLSと同じ情報・システム研究機構の組織ですので、DDBJと有機的に連携しながら開発を進めていければと考えています。
結果のランキングなど
RefSeq+INSDC全体をGGRNAに入れただけでは、おそらく検索ノイズが増えて使いにくくなってしまうと思われます。本当に使いやすいサービスにするためには、ユーザが求める情報に短時間で到達できるように検索結果の見せ方を工夫する必要があります。具体的には、ランキングを改良することと、結果を後から絞り込めるようにファセット検索を実装することを計画しています。
GGRNAのオープンソース化
論文投稿時にある方からGGRNAのオープンソース化を検討してはどうかと提案をいただきました。これは是非やってみたいです。エンジンは商用のプログラムを利用しているので、公開できるのはインターフェース部分が中心になりますが、ドキュメントをある程度整備したら公開する計画です。オープンに開発することによって、フィードバックを貰えたり、GGRNAを利用してもらう機会が増えるのは素晴らしいことだと思います。
Chateau Togo (by @iNut) より:
僕がこの牧場で教わったことは3つありますー
一つ、書いたコードは恥を承知で晒せ。
一つ、コマンドの使い方が分からなければmanを読め。
一つ、フォームを見たらとりあえずXSSを打ち込め。
- Comments (Close): 0
- Trackbacks: 0
GGRNA Search Examples (protein sequences)
- 2012-04-22 (日)
- DBCLS
Search for amino acids included in Figures
Not only nucleotide sequences, but also amino acid sequences appearing in article figures can be quickly searched using GGRNA.

[Schaefer et al. (1999) IV. Wilson’s disease and Menkes disease. Am. J. Physiol. Gastrointest. Liver Physiol. 276, G311-G314]
To search for the protein illustrated in the above figure, just enter partial amino acid sequence to the search box like [ aa:MTCQSC ] (→ GGRNA) or [ aa:MHCKSC ] (→ GGRNA) and you can get results. GGRNA works case insensitively, so [ AA:mhcksc ] will do the same.
GGRNA works great for not only searching genes, but also for locating the sequences. You can enter all the sequences shown in the figure like [ aa:MTCQSC aa:MHCKSC aa:MTCASC aa:CPC aa:DKTGT aa:SEHPL aa:GDGVND ] (→ GGRNA) to easily see where they are located.


Search caspase cleavage sites
Caspases are a group of cystein proteases involved in the induction of apoptosis, and they recognize certain peptide sequences to cleave proteins. By using GGRNA amino-acid search, you can find genes whose products are likely to be cleaved by capspases, and locate their cleavage sites. Shown below are the cleavage sites of caspase-3 and caspase-8. Note that GGRNA works in case insensitive manner, so [ aa:DEVD ] and [ AA:devd ] will return the same results.

In the amino-acid sequence displayed, you can see IETD with green highlight. Below that, the position corresponding to the query sequence is indicated as ‘AA_position 731.’ Please be aware that the number indicates the position of the first matched residue (in this case I), not the residue that gets cleaved (D).

Search for ER localization signals
The amino acid sequence ‘KDEL’ at the C-terminus serves as the endoplasmic reticulum (ER) retention signal. To search for the KDEL motif in GGRNA, enter [ aa:KDEL ] in the search box (→ GGRNA). An operator aa: restricts the search to within amino acid sequences only. Searching [ aa:KDEL ] in human will retrieve 359 results (as of RefSeq release 52, Mar. 2012), but these results contain KDELs that are not at the C-terminus.
On the other hand, transcripts annotated as GO:0005783 (endoplasmic reticulum) can be retrieved by searching [ GO:0005783 ] (→ GGRNA), which returns 1,985 results.
An intersection of these two searches can be obtained by entering the two keywords separated by a space: [ aa:KDEL GO:0005783 ] (→ GGRNA), which returns 28 results. Of these, 13 results contain the KDEL motif at the C-terminus. Searching by sequences and other keywords simultaneously is one of the unique advantages of GGRNA.

Search for plant peptide hormones
Search for plant peptide hormones using signal sequences (by @hkanekane).
- CLV3 → [ aa:PDPLHHH ] (→ Search for Arabidopsis thaliana genes using GGRNA)
- RGF → [ aa:PPRHN ] (→ Search for Arabidopsis thaliana genes using GGRNA)
For sequences this short, BLAST does not work very well. GGRNA does overwhelmingly better than BLAST in searching short sequences consisting of less than 5 amino acids, except that GGRNA cannot do fuzzy search. I’m planning to implement GGRNA with amino-acid fuzzy search near future.
- Comments (Close): 0
- Trackbacks: 0
GGRNA Search Examples (DNA sequences)
- 2012-04-22 (日)
- DBCLS
One of the strength of GGRNA is the ultrafast search of nucleotide and amino-acid sequences. Let’s learn how to search them.
In silico PCR: search for PCR primer binding sites
Have you ever thought “I just want to use the exact primers in this paper for PCR,” or “Oops I can’t remember which region my primers amplify?” Here’s how GGRNA can help.
Let’s say you want to use the following PCR primers:
Forward primer: CTAGCTGCCAAAGAAGGACAT
Reverse primer: CAATGAGATGTTGTCGTGCTC
Enter [ CTAGCTGCCAAAGAAGGACAT comp:CAATGAGATGTTGTCGTGCTC ] to the search box (→ GGRNA). Since reverse primer is designed against complementary sequence, add comp: operator to let GGRNA to search for complementary sequences.

In this case, two transcript variants of NFκB (NM_001165412.1, NM_003998.3) are retrieved. Let’s take a look at the first one (NM_001165412.1).
Look at the nucleotides highlighted with green: below the highlighted letters, you can see “position 2328 2547,” indicating the start positions of the sequences matched with the queried primers. You can calculate the size of the amplified product by using these numbers: 2547 – 2328 + 21 = 240 (bp), where 21 is the length of the reverse primer. You can also see (CDS: 468 – 3374) at the right side of “position,” which indicates the corresponding CDS region. It means that the primers used here are designed within CDS.
Now try to find out the length of the second (NM_003998.3) amplified products — yes, it’s 2550 – 2331 + 21 = 240 (bp)! This NFκB transcript variant seems to give products with the same size.
Let’s have a close look at the primer binding sites. Click the title of the first hit, “Homo sapiens nuclear factor of kappa …,” and the details of the amplified product are displayed, including the primer binding sites highlighted.

By the way, if you do the same search in UCSC In-Silico PCR, which is a famous and conventional web service to search for fragments amplified by the entered primers, it’ll come up with only one result with the size of 692 bp.

This discrepancy is attributable to the fact that UCSC service searches against genome, while GGRNA searches against transcripts. The above NFκB primers are designed to sandwich a 452 bp intron, so when you use genome DNA as a template, the product will be 240 + 452 = 692 (bp). When designing primers, sandwiching an intron is a good way: since the products derived from cDNA template and genomic DNA template will give different lengths, you can detect genomic DNA contamination in your template by simply looking at the product’s uniformity.
Search for nucleotide sequences included in figures
GGRNA works great for searching for nucleotide sequences appearing in figures of articles.

[Rajewsky et al. (2006) microRNA target predictions in animals. Nature Genetics 38, S8 – S13]
Left RNA strand shows the partial 3′ UTR sequence of myotrophin, which is the target of mice miR-375. Try searching with its partial sequence using GGRNA by selecting Mus musculus (mouse) and entering [ GUUGCAAGA ] to the search box (→ GGRNA). But it returns 322 hits, which is too much. So narrow them down by entering longer fragment [ GUUGCAAGAACAAA ] (→ GGRNA), and you’ll get 1 hit. Be aware that GGRNA treats U and T identically.

The sequence matched at position 3763, and CDS region is 279 – 635, meaning that miR-375 matches to farther downstream in 3′ UTR.
FYI, target sequence of miR-375 shown on the right side in the figure can be retrieved by entering about 13 letters, [ UUUGUUCGUUCGG ] (→ GGRNA).
Next.

[Yekta et al. (2004) MicroRNA-directed cleavage of HOXB8 mRNA. Science 304, 594-596]
Let’s search using the letters in black background [ CCAACAACAUGAAACUGCCUA ] (in human, mouse, or rat as you like) (→ GGRNA) and it’ll return position 1379 of HOXB8 (NM_024016.3) (CDS: 236 – 967), confirming that they indeed match to 3′ UTR.

For your information, search operators such as comp: for searching for complementary sequences, both: for searching for both strands, seq1:, seq2:, and seq3: for the search allowing 1, 2, or 3 mismatches respectively, are available.
Search for siRNA off-target transcripts
In mammalian RNAi, 21 nt double-stranded short interfering RNA (siRNA) is used. However, if an siRNA sequence resembles to another unrelated gene, it may unexpectedly suppress that gene. This is referred to as an off-target effect. In siDirect (website for designing functional siRNA with reduced off-target effect, which I have launched), designed siRNA sequences (19-mer sequence on the guide strand counting from 5′ positions 2 to 20) are used for homology search and it returns a list of genes homologous to the sequences (with up to 3 mismatches). By the way, why I use the 19-mer rather than the full length (positions 1 to 21) is that when RNAi takes place, the nucleotide at the 5′ end of the guide strand is in the pocket of Mid-domain in Argonaute protein, and the nucleotide at the 3′ end binds to PAZ domain, making them irrelevant for target mRNA recognition. The number of mismatches enough to avoid off-target effect has not been determined yet: 1 mismatch is definitely not enough, and as the number of mismatches increase, the risk of off-target effect decreases. From the bioinformatics analysis, it is possible to design siRNAs that have at least 3 mismatches to any other unrelated genes than the target sequence (for approximately 10% of the entire gene), but it is hardly possible to design them with at least 4 mismatches.
Here we use the below siRNA sequence to search for homologous genes using GGRNA. This siRNA is designed to target a gene called claudin 17.

What we want to do here is to search for sequences that hybridize with 5′-AGAACUUGCAUUGCAACCG-3’, which is the 19-mer of the siRNA guide strand 5′-UAGAACUUGCAUUGCAACCGG-3′ (both ends removed), so let’s start with entering [ comp:AGAACUUGCAUUGCAACCG ] to the search box (→ GGRNA).

Claudin 17 (CLDN17; NM_012131.2), the target gene of this siRNA is the only result retrieved. Now let’s try again with allowing mismatches.
- allows up to 1 mismatch → [ comp1:AGAACUUGCAUUGCAACCG ] (→ GGRNA)
- allows up to 2 mismatches → [ comp2:AGAACUUGCAUUGCAACCG ] (→ GGRNA)
- allows up to 3 mismatches → [ comp3:AGAACUUGCAUUGCAACCG ] (→ GGRNA)

When we tolerated 3 mismatches, 3 other hits are finally retrieved. siDirect will return the results shown below, and the difference between GGRNA and siDirect comes from the versions of the sequence database used (GGRNA uses newer version of the database). The mismatched positions are better visualized in siDirect than in GGRNA: I’m planning to update GGRNA to display the mismatched nucleotides in different color like siDirect near future.

Of course, passenger strand can cause off-target effect as well as guide strand, so the passenger strand should also be searched. From the passenger strand 5′-GGUUGCAAUGCAAGUUCUAUA-3′, remove the nucleotides at both ends and search for sequences hybridizing with 5′-GUUGCAAUGCAAGUUCUAU-3′:
- perfect match → [ comp:GUUGCAAUGCAAGUUCUAU ] (→ GGRNA): no hit
- up to 1 mismatch → [ comp1:GUUGCAAUGCAAGUUCUAU ] (→ GGRNA): no hit
- up to 2 mismatches → [ comp2:GUUGCAAUGCAAGUUCUAU ] (→ GGRNA): no hit
- up to 3 mismatches → [ comp3:GUUGCAAUGCAAGUUCUAU ] (→ GGRNA): 5 hits
Here 3 mismatches had to be tolerated to return off-target candidates.
It is very rare to have few hits when you search 19-mer with 3 mismatches allowed. Thus the siRNA in the above example seems highly specific in terms of nucleotide sequence.
By the way, ‘seed’ sequences (7-mer nucleotides in positions 2 to 8 of guide strand) with higher Tm also have the risk of exerting off-target effect to genes whose 3′ UTR contain sequences identical to the seed. Please refer to the below paper or TogoTV guide for siDirect for further information. To design siRNAs with less off-target effect, you have to start with choosing sequences with lower seed Tm.
- Naito et al. (2009) siDirect 2.0: updated software for designing functional siRNA with reduced seed-dependent off-target effect. BMC Bioinformatics 10, 392 → full text
- TogoTV: Designing siRNA with siDirect: 2011 (in Japanese only)
Search for nucleotide sequences using microarray probe ID
As I have shown in the entry on 2/Jun/2011 “Nucleotide search using microarry probe ID” (in Japanese), when you enter microarray probe IDs, GGRNA will search for genes using the nucleotide sequences corresponding to the probe IDs. It also specifies the positions where probes hybridize.
In particular, Affymetrix microarray uses eleven 25-mer perfect match (PM) probes, collectively called ‘probeset,’ to recognize single transcript. As shown below, Affymetrix also provides mismatch (MM) probes at the same positions as the 11 PM probes for background, but it is not used as often as before.

When probeset ID is entered in GGRNA like [ 1552311_a_at ] (→ GGRNA), GGRNA converts probeset ID into nucleotide sequences to perform search with:
[ GCATGGGATGGGACAGTCTGGGCCA ] +
[ AGAAGTGCGGCACCAGGGCAGGAGC ] +
[ GGCAGGAGCTGCAGTAGCTACCCTC ] +
[ AGATCACTCCCAGATCACCAGGTCA ] +
[ AGGTCACCCCATCTCTAGGCGGCAC ] +
[ AATGTCACCGCACACCAGGCAGTGG ] +
[ GGGACACGGCAGTAAGCACAAGAAA ] +
[ ACGGCAGTAAGCACAAGAAAGATTT ] +
[ TCTCCACAAACGTTTTTAAAATGTG ] +
[ AAAATGTGCCGGGTGTACTGGTGCA ] +
[ ATGTGCCGGGTGTACTGGTGCACAC ] .

The search returns RAX2 (NM_032753) gene. Click the title of the hit…

…and you will see that the 11 oligonucleotides hybridize with sequences close to 3′ end. Nucleotides with overlapping hits are shown in dark green.
Meanwhile, Agilent microarray uses one 60-mer probe to recognize single transcript. For example, searching with [ A_23_P101434 ] will return the following result (→ GGRNA):

Search for binding motifs of RNA-binding protein
Degenerate motifs recoognized by RNA binding proteins can be searched using IUB codes (e.g., N, R, Y). For example, search for mRNA having PUM binding site UGUANAUA by entering [ iub:UGUANAUA ] to the search box (→ GGRNA: will take about 10 secs).

The search returns 9,720 hits. You can narrow them down by entering other keywords, or use the tab-delimited format provided at the bottom of the page for further analyses using other softwares.

- Comments (Close): 0
- Trackbacks: 0
統合TVにGGRNAの解説が登場!
- 2012-01-24 (火)
- DBCLS
統合TV:GGRNAで遺伝子をGoogleのように検索する
ついに統合TVにGGRNAが登場しました! 制作は @Masamichi_C2 さん。動画は8分近くありますが、基本操作の紹介から活用事例まで盛り込んで作ってくれたので、ぜひご覧ください。ところで、今回の統合TVからBGMが入りました(笑)。近いうちに音声の解説が入った番組も公開できるよう、統合TVチームが準備を進めています。

- Comments (Close): 0
- Trackbacks: 0
- Search
- Feeds
- Meta